
MDCG 2020-16 rev.1: guida alle regole di classificazione dei Dispositivi Medici Diagnostici in vitro (IVD)
MDCG 2020-16 rev.1: Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746
La MDCG 2020-1 è molto utile per chiarire il processo di assegnazione della classe di rischio dei Dispositivi Medici Diagnostici in vitro (IVD).
Questa MDCG è essenziale non solo per i fabbricanti di IVD ma anche per gli Organismi Notificati in quanto riporta un metodo e degli esempi utili per uniformare l’implementazione dell’annex VIII del Regolamento (UE) 2017/746 (IVDR).
Da questa MDCG è stata ribadita l’importanza di scegliere e definire con cura lo scopo e la destinazione d’uso del dispositivo medico diagnostico in vitro per poi determinare con facilità la classe di rischio.
Per gli IVD le classi di rischio sono 4 e sono identificate con le prime 4 lettere dell’alfabeto: la A è la classe di rischio più bassa e la D è la classe di rischio più alta. Per determinare la classe di rischio cui un dispositivo appartiene, l’annex VIII del regolamento (UE) 2017/746 (IVDR) riporta 7 regole di classificazione, che riportiamo nel seguito per completezza.
Determinare correttamente la classe di rischio del proprio Dispositivo Medico Diagnostico in Vitro è essenziale in quanto ogni classe ha degli obblighi differenti, legati al rischio stesso del prodotto.
Uno dei requisiti la cui implementazione varia al variare della classe di rischio è l’obbligatorietà dell’apposizione dell’UDI sull’etichetta in quanto, come descritto nell’articolo 113 del Regolamento, sarà introdotto gradualmente con i seguenti tempi:
- Classe D: 26 maggio 2023
- Classe B e C: 26 maggio 2025
- Classe A: 26 maggio 2027
Un altro esempio dell’importanza di definire con esattezza la classe di rischio del vostro IVD riguarda la sezione del regolamento che tratta della “valutazione della conformità”, come specificato nell’art 48.
Di seguito una sintesi dell’articolo:
- Classe D Il fabbricante può scegliere una tra le seguenti opzioni:
- Allegato IX, capitoli I, II (ad eccezione della sezione 5) e III
- Allegato X insieme all’Allegato XI
- Classe C Il fabbricante può scegliere tra le opzioni:
- Allegato IX, Capitoli I e III (compresa una valutazione della documentazione tecnica di almeno un dispositivo rappresentativo per gruppo di dispositivi generico)
- Allegato IX, Capitoli I, II e III che include la sezione 5 in cui il dispositivo sia un self-test, un test diagnostico per analisi decentrate o un test di accompagnamento
- Allegato X insieme all’Allegato XI
- Classe B Il fabbricante può scegliere tra le opzioni:
- Allegato IX, capitoli I e III (compresa una valutazione della documentazione tecnica di almeno un dispositivo rappresentativo per categoria di dispositivi)
- Allegato IX, Capitoli I, II e III, che include la sezione 5 in cui il dispositivo sia un self-test, un test diagnostico per analisi decentrate
- Classe A:
Autodichiarazione a meno che i dispositivi non siano immessi sul mercato in condizioni sterili, nel qual caso il fabbricante applicherà gli allegati IX o XI.
Per i dispositivi di classe B, C e D sarà necessaria la valutazione di conformità da parte di un Organismo Notificato.
NB: La classificazione di un dispositivo è in primo luogo responsabilità del fabbricante. Se l’Organismo Notificato non è d’accordo con la classificazione del fabbricante, la questione viene sottoposta all’Autorità Competente del Paese in cui ha sede il fabbricante (o il suo mandatario, se il fabbricante ha sede fuori dall’Unione Europea).
Se l’Organismo Notificato e il Fabbricante hanno sede in due Paesi diversi, l’Autorità Competente del Paese in cui ha sede il Fabbricante prende una decisione solo dopo aver consultato l’Autorità Competente del Paese che ha designato l’Organismo Notificato.

Figura 1 Alcuni requisiti dipendenti dalla classe di rischio
Di seguito si riportano delle tabelle riassuntive che hanno lo scopo di facilitare il processo di assegnazione della classe di rischio ai Dispositivi Medici Diagnostici in Vitro:
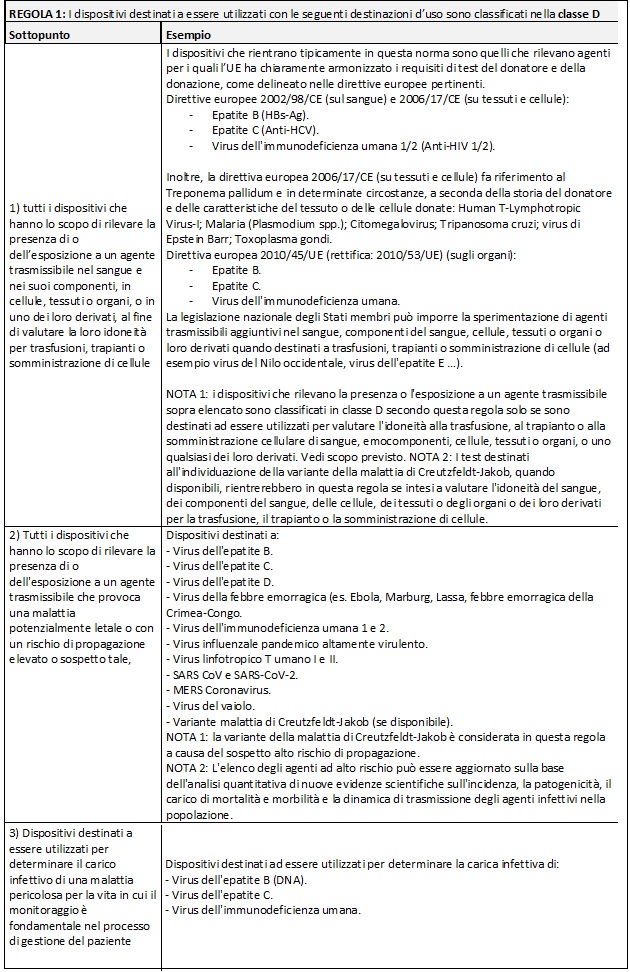

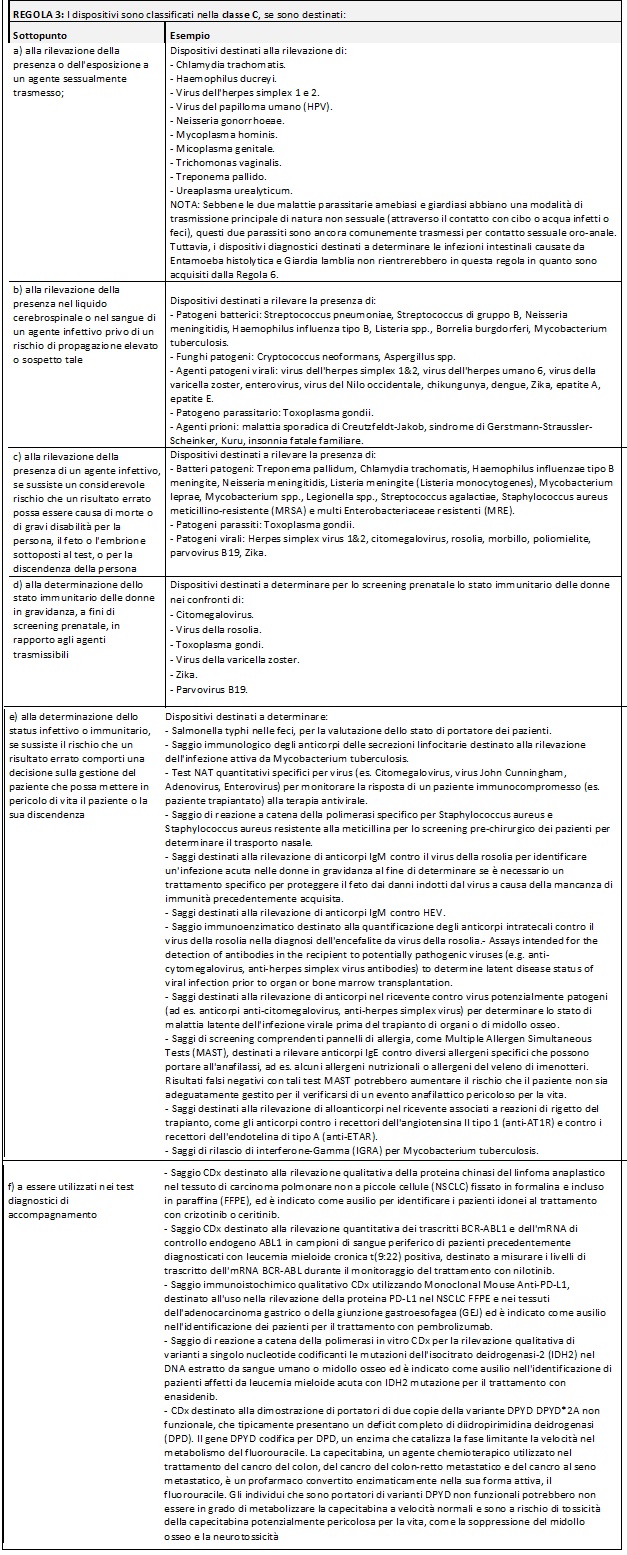

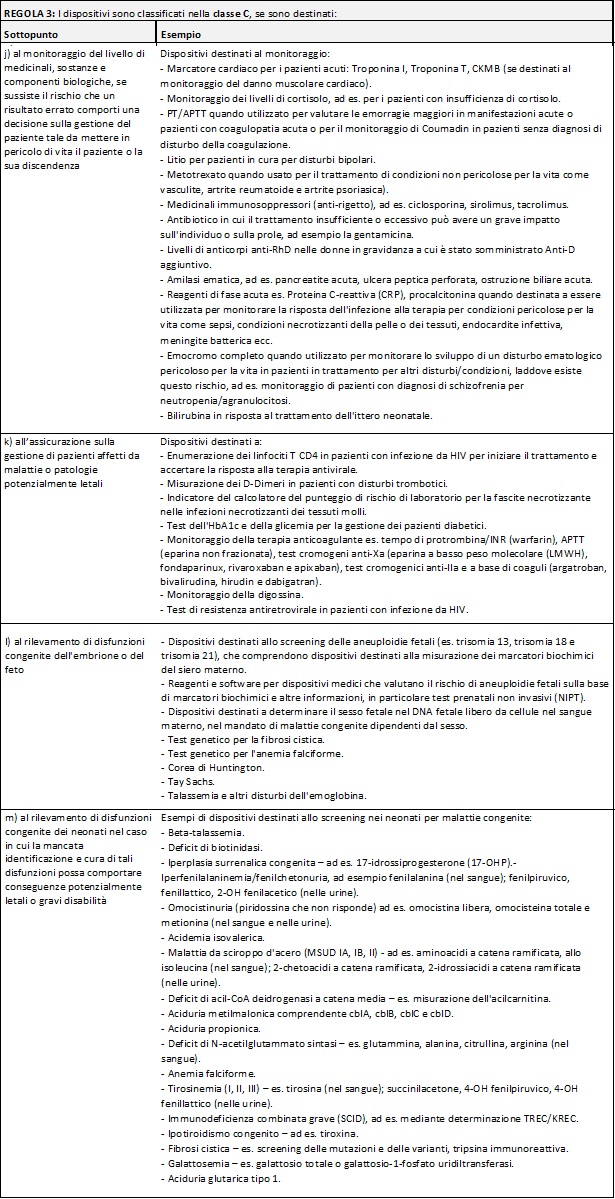
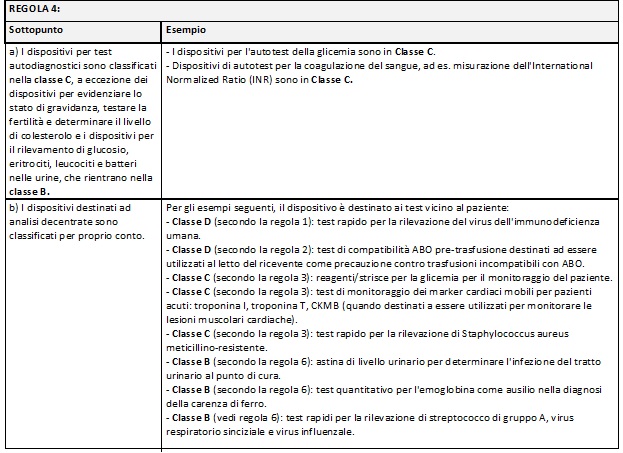
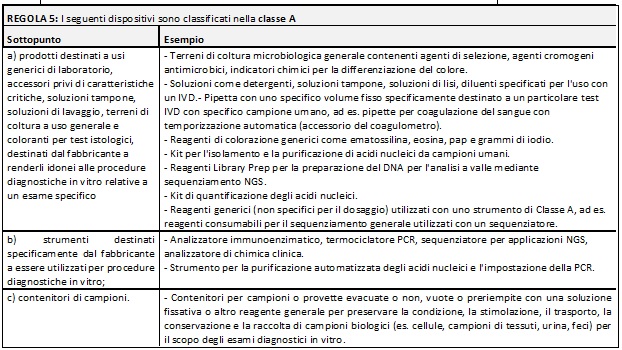
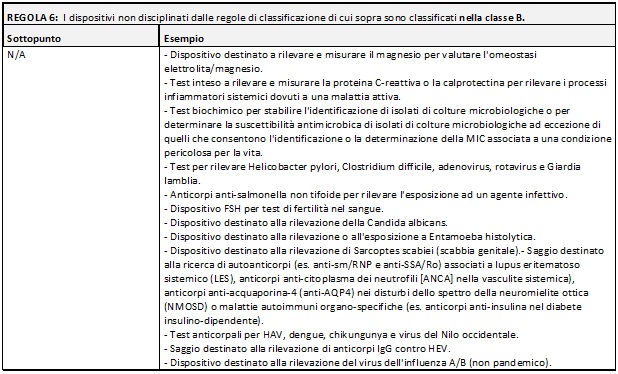

Nella MDCG potete trovare i razionali dove la commissione delinea la ragione di ogni singola regola e relativi sottopunti.
Personalmente mi ha stupito la volontà della commissione nel dare dei razionali ed esempi per ogni regola di classificazione, in modo da avere sempre meno dispositivi border-line che potrebbero essere definiti in più regole. Ovviamente in caso ci fosse una situazione simile e nemmeno la MDCG ha risolto i vostri dubbi, usate la regola che prevede la classificazione più elevata ma prima contattate il vostro consulente e/o project handler del vostro organismo notificato.
Se non avete ancora un consulente di fiducia, scriveteci a info@mediconsulting.eu e saremo ben lieti di supportarvi nel Vostro percorso.